Pharmaceutical Cleanroom Design: TGA Compliance and GMP Requirements
A pharmaceutical cleanroom in Australia is a controlled environment designed to meet TGA Good Manufacturing Practice (GMP) requirements for the manufacture of sterile or non-sterile pharmaceutical products. Cleanrooms are classified by air quality from Grade A (ISO 5 - used for aseptic filling operations) through to Grade D (ISO 8, for lower-risk manufacturing activities). Each classification determines the required air changes per hour, particulate limits, pressure differentials, filtration specifications, and monitoring requirements under Australian regulations.
In the highly regulated world of pharmaceutical manufacturing, cleanroom design is not merely a technical consideration - it's a critical foundation for product quality, patient safety, and business success. For pharmacy owners and business leaders looking to establish or upgrade sterile manufacturing facilities in Australia, navigating the complex landscape of Therapeutic Goods Administration (TGA) requirements and Good Manufacturing Practice (GMP) standards can be daunting.
A well-designed pharmaceutical cleanroom does more than simply meet regulatory requirements; it creates an environment where contamination risks are minimised, operational efficiency is maximised, and compliance is built into the very structure of your facility. This is particularly crucial for sterile manufacturing processes, where even minor design oversights can lead to significant product quality issues, failed inspections, or costly remediation efforts.
This comprehensive guide explores the essential elements of pharmaceutical cleanroom design in Australia, with a specific focus on meeting TGA compliance requirements and GMP standards for sterile manufacturing facilities.
Understanding Cleanroom Classifications for Pharmaceutical Manufacturing
Before embarking on any cleanroom design project, it's essential to understand the classification system that governs these specialised environments and how Australian requirements align with international standards.
Australian Cleanroom Standards and ISO Classifications
In Australia, pharmaceutical cleanrooms must comply with both international standards and specific TGA requirements. Pharmacy compounding operations fall under a separate regulatory framework from manufacturing cleanrooms - if you are designing a compounding space rather than a full manufacturing facility, our compounding lab design guide covers the workflow, cleanroom classifications, and TGA and Pharmacy Board requirements that apply specifically to pharmacy compounding. The primary reference for cleanroom classification is ISO 14644-1, which defines cleanrooms based on the concentration of airborne particles.
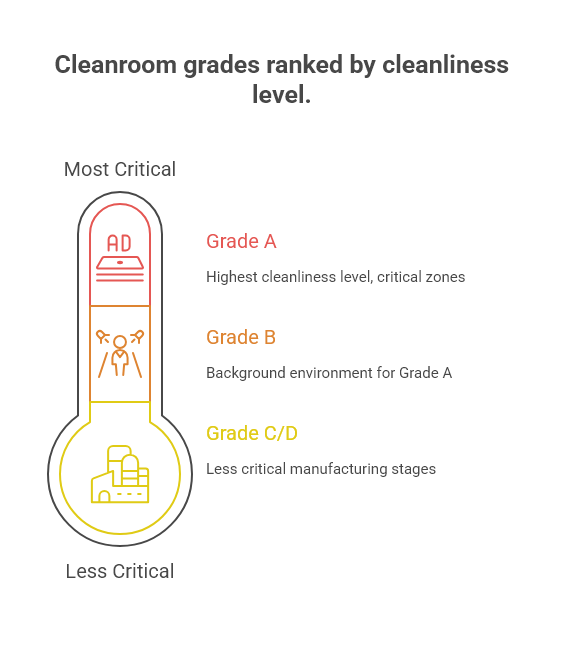
The TGA, through its adoption of the Pharmaceutical Inspection Co-operation Scheme (PIC/S) Guide to Good Manufacturing Practice, recognises four main grades of cleanrooms for pharmaceutical manufacturing:
Grade A: The highest cleanliness level, typically used for critical zones where products, containers, and closures are exposed to the environment (e.g., filling zones, stopper bowls).
Grade B: The background environment for Grade A zones in aseptic preparation and filling.
Grade C and D: Clean areas for carrying out less critical stages of manufacturing.
According to the TGA's interpretation of PIC/S Annex 1, these grades correspond to specific maximum permitted airborne particle concentrations. For example, Grade A environments must maintain ≤3,520 particles/m³ at ≥0.5μm and ≤20 particles/m³ at ≥5.0μm when measured in the "at rest" state (TGA, 2023).
Beyond particle counts, Australian cleanroom standards also specify requirements for:
Air changes per hour: Grade A/B areas typically require 15-20 air changes per hour, while Grade C may require 10-15 and Grade D 5-10, depending on room size and heat load.
Pressure differentials: A minimum of 10-15 Pascal pressure differential between adjacent rooms of different classifications, with a cascade from highest to lowest grade.
Temperature and humidity control: Typically 18-22°C and 30-65% relative humidity, though specific products may require tighter controls.
Selecting the Appropriate Classification for Your Manufacturing Process
Determining the correct cleanroom classification is a critical first step that will influence virtually every subsequent design decision. The Australian regulatory framework requires that this determination be based on a documented risk assessment of your specific manufacturing process.
For sterile manufacturing, the TGA expects a comprehensive contamination control strategy that justifies your cleanroom classification choices based on:
Product characteristics: Injectable products generally require the highest cleanliness standards.
Manufacturing process: Terminal sterilisation allows for somewhat lower environmental controls compared to aseptic processing.
Exposure risk: The degree and duration of product exposure to the environment.
According to the Pharmaceutical Society of Australia's guidelines, sterile compounding facilities must conduct a formal risk assessment to determine appropriate cleanroom classifications based on the complexity of preparations and potential impact on patient safety (PSA, 2022).
Key TGA Compliance Requirements for Pharmaceutical Cleanrooms
Achieving and maintaining TGA compliance requires a thorough understanding of both explicit requirements and regulatory expectations for pharmaceutical manufacturing facilities.
PIC/S Guide to GMP and TGA Interpretations
The TGA has adopted the PIC/S Guide to Good Manufacturing Practice as the standard for pharmaceutical manufacturing in Australia. Particularly relevant to cleanroom design is Annex 1, which covers the manufacture of sterile medicinal products.
Key TGA interpretations of PIC/S requirements that impact cleanroom design include:
Contamination control strategy: The TGA now requires a holistic, documented approach to contamination control that encompasses facility design, equipment, utilities, and personnel.
Quality risk management: All design decisions must be supported by formal risk assessments following ICH Q9 principles.
Continuous monitoring: Modern cleanrooms are expected to incorporate real-time monitoring systems rather than relying solely on periodic testing.
The Australian regulatory framework places particular emphasis on the concept of "design for quality," where compliance is built into the facility rather than tested into the product. According to the TGA's guidance on GMP clearance for overseas manufacturers, facilities designed with compliance as a foundational principle are more likely to receive approval and maintain it through ongoing inspections (TGA, 2023).
Critical Design Elements for TGA Approval
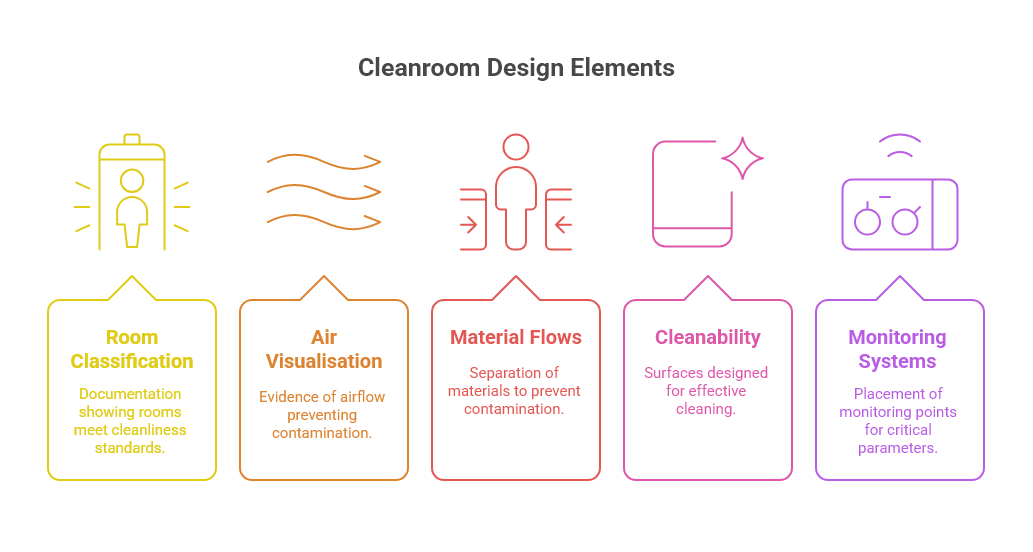
TGA inspectors focus on several key design elements when assessing pharmaceutical cleanrooms:
Room classification and qualification: Documentation demonstrating that rooms meet their intended cleanliness classification under both "at rest" and "in operation" conditions.
Air visualisation studies: Evidence that airflow patterns prevent cross-contamination and maintain product protection.
Material and personnel flows: Clear separation of materials, personnel, and waste to prevent cross-contamination.
Cleanability: Surfaces and interfaces designed for effective cleaning and disinfection.
Monitoring systems: Appropriate placement of monitoring points for viable and non-viable particles, pressure differentials, and other critical parameters.
The Australian Commission on Safety and Quality in Health Care notes that facilities designed with these elements in mind from the outset face fewer compliance challenges and remediation costs compared to those that attempt to retrofit compliance measures after construction (ACSQHC, 2022).
Essential Components of Pharmaceutical Cleanroom Design
A compliant pharmaceutical cleanroom is a complex system where multiple components must work together seamlessly to maintain the required environmental conditions.
HVAC Systems and Air Handling Requirements
The heating, ventilation, and air conditioning (HVAC) system is perhaps the most critical element of cleanroom design, responsible for maintaining appropriate air quality, temperature, humidity, and pressure differentials.
For Australian pharmaceutical cleanrooms, key HVAC design considerations include:
HEPA filtration: Terminal High-Efficiency Particulate Air (HEPA) filters are mandatory for Grade A/B areas and recommended for Grade C. The Australian Institute of Refrigeration, Air Conditioning and Heating (AIRAH) recommends minimum H14 class HEPA filters (99.995% efficient at MPPS) for critical applications (AIRAH, 2023).
Air distribution patterns: Unidirectional (laminar) airflow is required for Grade A zones, while non-unidirectional airflow may be acceptable for lower grades. The design must demonstrate appropriate air distribution through computational fluid dynamics (CFD) modelling or smoke studies.
Energy efficiency: Given Australia's focus on energy conservation, modern cleanroom HVAC systems should incorporate energy recovery systems, variable frequency drives, and smart controls while still meeting GMP requirements. The Green Building Council of Australia notes that pharmaceutical facilities can reduce energy consumption by 20-30% through optimised HVAC design without compromising compliance (GBCA, 2023).
Redundancy: Critical systems require appropriate redundancy to ensure continuous operation during maintenance or component failure.
Material Selection for Walls, Floors, and Ceilings
The materials used in cleanroom construction must meet stringent requirements for cleanability, durability, and particle generation while complying with Australian building codes.
Recommended materials for Australian pharmaceutical cleanrooms include:
Walls: Pharmaceutical-grade panels with welded seams or monolithic epoxy-coated plasterboard. All joints should be coved and sealed to eliminate crevices where contamination can accumulate.
Floors: Seamless vinyl or epoxy flooring that curves up the walls to create coved bases. The National Construction Code of Australia requires that flooring materials in healthcare facilities meet specific slip resistance ratings (ABCB, 2022).
Ceilings: Walkable or non-walkable sealed panel systems with gasketed HEPA filter housings. Lighting fixtures should be flush-mounted and sealed.
Doors and windows: Flush-mounted with minimal ledges and appropriate seals. Windows between classified areas should be sealed units.
According to Standards Australia, materials used in pharmaceutical cleanrooms should be tested for particle shedding, chemical resistance, and microbial growth resistance to ensure long-term compliance with cleanliness requirements (Standards Australia, 2023).
Airlocks, Pass-Throughs, and Personnel Flows
Transition spaces are critical for maintaining cleanroom integrity and preventing cross-contamination.
Key design considerations include:
Personnel airlocks: Should include sufficient space for gowning/de-gowning and hand hygiene facilities. The TGA expects separate entry and exit airlocks for the highest grade areas to prevent cross-contamination.
Material airlocks: Should be designed with appropriate size for the materials being transferred and may require transfer hatches or pass-through chambers with interlocking doors.
Pass-through hatches: Should maintain pressure differentials and may include UV sanitisation or HEPA-filtered air showers depending on the application.
Unidirectional flow: The facility layout should enforce a logical progression from lower to higher classification areas, with no backtracking that could introduce contamination.
The Australasian Health Infrastructure Alliance guidelines recommend that cleanroom designs include clear delineation of personnel and material flows with physical barriers where possible to prevent cross-contamination (AHIA, 2022).
Contamination Control Strategies in Sterile Manufacturing Environments
Effective contamination control requires a holistic approach that considers all potential sources of contamination and implements appropriate barriers.
Personnel and Material Movement Planning
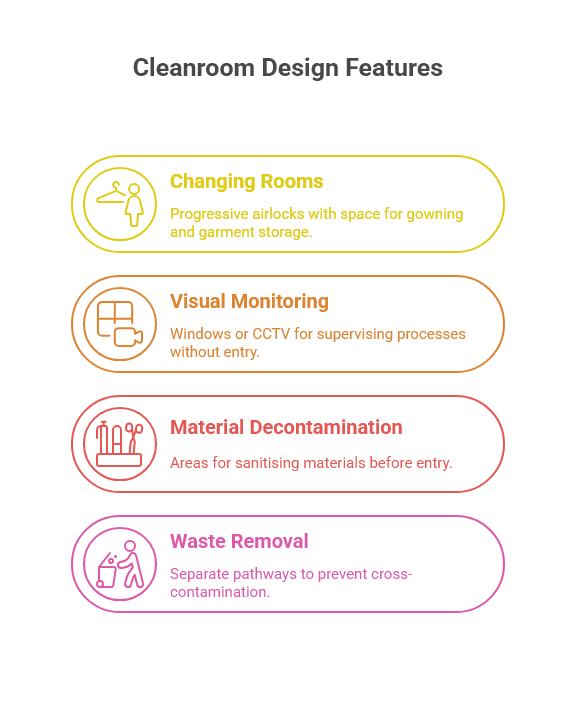
People are typically the greatest source of contamination in cleanrooms. A well-designed facility minimises this risk through thoughtful workflow planning:
Changing rooms and gowning areas: Should be designed as progressive airlocks with appropriate space for gowning procedures and storage of clean garments.
Visual monitoring: Windows or CCTV systems that allow supervision of critical processes without additional personnel entering the cleanroom.
Material decontamination: Dedicated areas for sanitising materials before entry into classified areas, potentially including automated systems for high-volume items.
Waste removal: Separate pathways for waste removal that prevent cross-contamination with incoming materials or finished products.
The pharmaceutical manufacturing industry recommends that facility designs include detailed personnel and material flow diagrams that are validated through simulation before construction begins.
Environmental Monitoring Systems and Integration
Modern pharmaceutical cleanrooms require comprehensive monitoring systems that provide both real-time data and historical records for trend analysis:
Particle counters: Fixed or portable systems for monitoring non-viable particles, strategically placed at critical locations.
Microbial sampling points: Designated locations for active and passive microbial monitoring.
Pressure, temperature, and humidity sensors: Continuous monitoring with appropriate alarm systems for out-of-specification conditions.
Building management system integration: All monitoring systems should feed into a central building management system (BMS) with appropriate data storage and reporting capabilities.
The National Association of Testing Authorities, Australia (NATA) provides guidance on the validation of environmental monitoring systems, emphasising the importance of appropriate sensor placement and calibration procedures (NATA, 2023).
Utilities and Support Systems for Pharmaceutical Cleanrooms
The utility systems supporting a pharmaceutical cleanroom are as critical as the cleanroom itself and must meet the same high standards for design and validation.
Water Systems for Pharmaceutical Manufacturing
Water is a primary ingredient in many pharmaceutical products and a critical utility for cleaning processes. Australian cleanroom facilities typically require:
Purified Water (PW) system: Meeting the requirements of the British Pharmacopoeia or European Pharmacopoeia, typically produced through reverse osmosis and/or deionisation.
Water for Injection (WFI) system: For parenteral products, meeting stricter requirements and typically produced through distillation or validated reverse osmosis in Australia.
Distribution systems: Designed to prevent microbial growth through continuous circulation, elevated temperatures, regular sanitisation, and point-of-use filtration.
The TGA's guidance on water quality for pharmaceutical manufacturing emphasises the need for materials that prevent contamination, such as electropolished stainless steel (316L) for distribution piping and appropriate sanitisation ports (TGA, 2022).
Process Gases and Compressed Air Systems
Clean gases and compressed air are essential utilities in many pharmaceutical processes:
Compressed air: Medical-grade compressed air systems require oil-free compressors, appropriate filtration, and regular monitoring for viable and non-viable contaminants.
Process gases: Nitrogen, oxygen, and other process gases require appropriate purity certification and point-of-use filtration.
Distribution systems: Should be constructed from appropriate materials (typically 316L stainless steel or pharmaceutical-grade plastics) with minimal dead legs and appropriate monitoring points.
Standards Australia provides specific guidance on medical gas pipeline systems that applies to pharmaceutical manufacturing facilities, covering materials, installation, and validation requirements (Standards Australia, 2022).
Validation and Qualification of Cleanroom Facilities
Validation is not an afterthought but an integral part of the design process for pharmaceutical cleanrooms in Australia.
Design Qualification and User Requirement Specifications
The validation process begins with clear documentation of requirements:
User Requirement Specifications (URS): Should detail all functional, operational, and compliance requirements for the facility.
Design Qualification (DQ): Formal verification that the proposed design meets all requirements specified in the URS and complies with GMP principles.
Risk assessments: Documented analysis of potential failure modes and their impact on product quality and patient safety.
The TGA expects to see evidence that validation was considered from the earliest design stages, with appropriate subject matter experts involved in reviewing designs before construction begins (TGA, 2023).
Installation, Operational, and Performance Qualification
Once construction is complete, a rigorous qualification process verifies that the facility performs as intended:
Installation Qualification (IQ): Verifies that all systems are installed correctly according to approved specifications.
Operational Qualification (OQ): Confirms that systems operate as intended across normal and challenge conditions.
Performance Qualification (PQ): Demonstrates that the facility consistently performs as required during simulated or actual production.
The Australian pharmaceutical bioprocessing industry recommends that qualification protocols include worst-case scenarios and challenge tests to ensure robust performance under all conditions (Monash University, 2023).
Cost Considerations and Return on Investment
While compliance is non-negotiable, there are ways to optimise the cost-effectiveness of pharmaceutical cleanroom projects.
Initial Design vs. Operational Costs
A well-planned cleanroom balances initial construction costs with long-term operational expenses:
Energy efficiency: Higher upfront investment in efficient HVAC systems and controls can reduce operational costs significantly over the facility lifecycle. The Australian Energy Efficiency Council estimates that pharmaceutical cleanrooms can reduce energy consumption by 25-40% through optimised design without compromising performance (EEC, 2023).
Maintenance accessibility: Designing for easy maintenance access to filters, motors, and other components reduces downtime and maintenance costs.
Material selection: Higher-quality materials may have greater upfront costs but offer longer lifespans and better cleanability, reducing long-term expenses.
Automation: Automated monitoring and control systems represent a significant initial investment but can reduce labour costs and compliance risks over time.
Scalability and Future-Proofing Your Cleanroom Investment
Given the substantial investment required for pharmaceutical cleanrooms, designing for future flexibility is essential:
Modular design: Using modular construction techniques allows for easier future expansion or reconfiguration.
Oversized utilities: Designing utility systems with capacity for future expansion can avoid costly retrofits later.
Adaptable classification: Designing areas that can be upgraded to higher classifications if needed through additional filtration or monitoring.
Technology readiness: Including infrastructure for future technology integration, such as conduit paths for additional monitoring systems or automation.
The Australian Manufacturing Growth Centre notes that pharmaceutical manufacturers who design facilities with future flexibility in mind typically achieve 15-20% lower lifetime costs compared to those requiring major retrofits for expansion (AMGC, 2023).
Working with Design Specialists: The Collaborative Approach
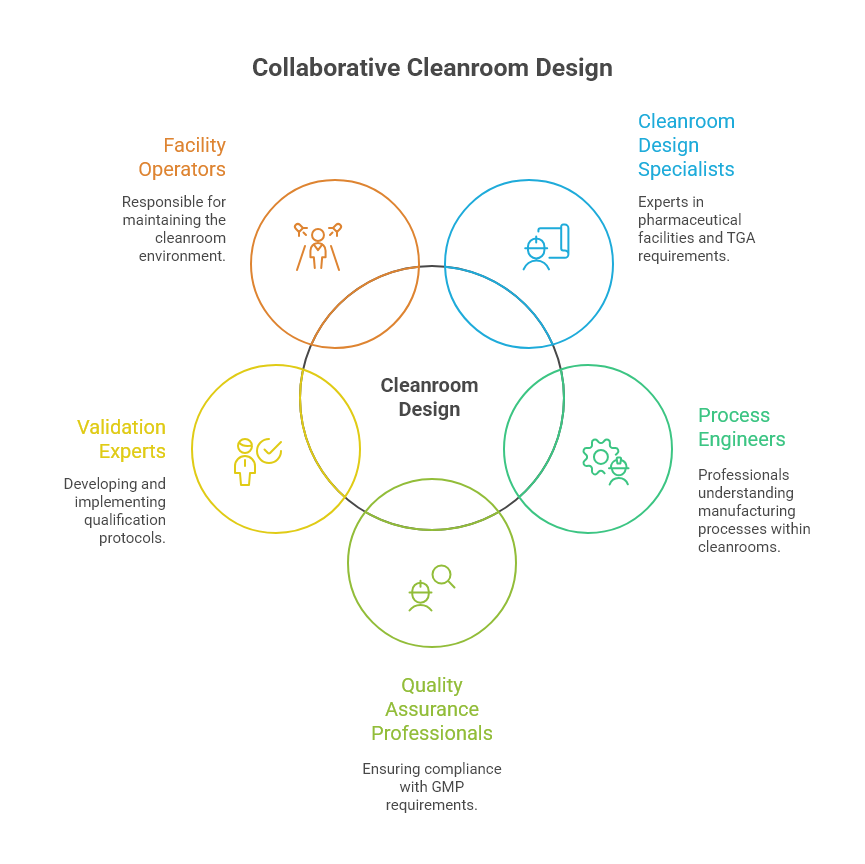
The complexity of pharmaceutical cleanroom design necessitates a collaborative approach involving multiple specialists.
A successful cleanroom project typically involves:
Cleanroom design specialists: With specific experience in pharmaceutical facilities and TGA requirements.
Process engineers: Who understand the manufacturing processes that will occur within the cleanroom.
Quality assurance professionals: To ensure compliance with GMP requirements throughout the design process.
Validation experts: To develop and implement appropriate qualification protocols.
Facility operators: Who will ultimately be responsible for maintaining the cleanroom environment.
At Design Yard 32, we understand that pharmaceutical cleanroom design requires this collaborative approach. Our commercial design services bring together the necessary expertise to create facilities that not only meet regulatory requirements but also support efficient operations and future growth.
Our team works closely with pharmaceutical clients to understand their specific processes and compliance needs, developing tailored solutions that balance regulatory requirements with practical operational considerations. Through our collaborative design process, we ensure that all stakeholders have input into the design from the earliest stages, preventing costly changes during construction or operation.
For pharmacy owners looking to establish or upgrade pharmaceutical manufacturing facilities, our pharmaceutical manufacturing and cleanroom design services provide comprehensive support from initial concept through to TGA licensing. Working in close collaboration with experienced GMP and ISO compliance consultants, we deliver facility designs that enable you to obtain and maintain TGA licensing while optimising workflow and operational efficiency.
-
At minimum, sterile compounding facilities must include appropriately classified clean zones (typically Grade A processing areas with Grade B backgrounds for aseptic processing), airlocks for personnel and materials, HEPA-filtered air supply, appropriate pressure differentials, and monitoring systems. The Pharmacy Board of Australia requires that facilities comply with the relevant sections of the PIC/S Guide to Good Manufacturing Practice and the Australian standard for cleanrooms (Pharmacy Board of Australia, 2023). The specific requirements depend on the risk level of the products being manufactured.
-
For a moderate-sized pharmaceutical cleanroom facility in Australia, the timeline typically includes 3-6 months for design and engineering, 1-2 months for regulatory review and approvals, 6-12 months for construction, and 2-3 months for commissioning and qualification. The Australian Construction Industry Forum notes that pharmaceutical projects often experience longer timelines than other construction projects due to the rigorous documentation and testing requirements (ACIF, 2023). Early engagement with regulatory consultants can help streamline the approval process.
-
According to TGA inspection reports, the most common design-related deficiencies include inadequate separation of personnel and material flows, insufficient pressure differentials between adjacent spaces, improper surface finishes that hinder effective cleaning, inadequate monitoring systems, and HVAC systems that cannot maintain required conditions under full heat load. The TGA's annual inspection deficiency report indicates that approximately 30% of GMP deficiencies relate to premises and equipment issues (TGA, 2023). Addressing these issues during the design phase is significantly more cost-effective than remediation after construction.
-
Energy-efficient cleanroom design strategies that maintain compliance include variable air volume systems that adjust air changes based on occupancy or production status, energy recovery systems that capture waste heat, LED lighting with occupancy sensors, and building management systems that optimise operations. The Green Building Council of Australia has documented case studies where pharmaceutical facilities reduced energy consumption by 30-40% through these approaches while maintaining or improving compliance standards (GBCA, 2023). The key is to thoroughly validate that energy-saving measures do not compromise the cleanroom's ability to maintain required conditions under all operating scenarios.
-
Before engaging design specialists, prepare a detailed user requirement specification (URS) that includes product information, batch sizes and frequencies, process descriptions, equipment lists with utilities requirements, personnel numbers, material flows, and specific regulatory requirements that apply to your products. The International Society for Pharmaceutical Engineering (ISPE) recommends also developing a preliminary contamination control strategy and quality risk assessment to guide the design process (ISPE, 2023). These documents help design specialists understand your needs and develop appropriate solutions from the outset, reducing the need for costly design changes later.